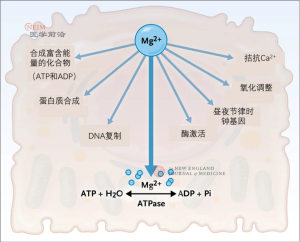

镁的调控器官主要有三个:肠道(调节膳食镁吸收)、骨骼(将镁以羟基磷灰石的形式储存)和肾脏(调节尿镁排泄)。这些系统相互整合、高度协调,共同构成肠道-骨骼-肾脏轴,分别负责镁的吸收、交换和排泄。镁代谢平衡失调可能导致病理生理结局。
镁是骨骼的重要组成部分,人体内60%的镁储存在骨骼中。骨骼中可交换镁为维持血浆生理浓度提供了动态储备。镁通过影响成骨细胞和破骨细胞活性促进骨骼形成。增加镁摄入量可提高骨骼矿物质含量,从而降低衰老过程中骨折和骨质疏松症风险。镁在骨骼修复中具有双重作用。在炎症急性期,镁可促进巨噬细胞中TRPM7表达、镁依赖性细胞因子产生以及促成骨形成的免疫微环境。在骨骼愈合的后期重塑阶段,镁可影响成骨并抑制羟基磷灰石沉淀。TRPM7和镁还通过影响血管平滑肌细胞向成骨表型转换而参与血管钙化过程。
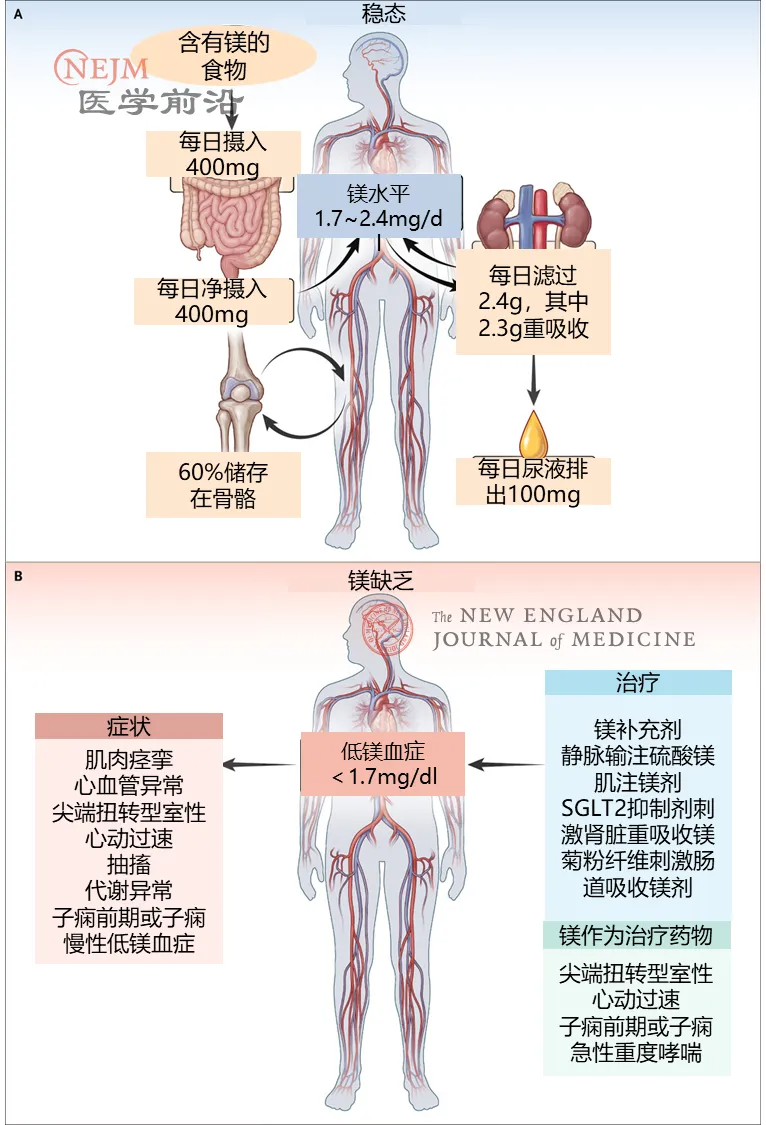
成人正常血清镁浓度为1.7~2.4 mg/dl(0.7~1.0 mmol/L)。低镁血症指血清镁浓度低于1.7 mg/dl。大多数交界性低镁血症患者无明显症状。由于在血清镁>1.5 mg/dl(0.6 mmol/L)时患者可能仍存在长期潜在镁缺乏的症状,因此有人建议将低镁血症的下限阈值提高。然而,这一水平仍存在争议,需要进一步临床验证。3%~10%的普通人群存在低镁血症,而2型糖尿病患者(10%~30%)和住院患者(10%~60%)的发病率更高,尤其是重症监护室(ICU)患者,其发病率超过65%。多个队列研究表明,低镁血症与全因死亡风险增加及心血管疾病相关死亡风险增加有关。
血液中的镁含量占比不到1%,因此血镁含量不能可靠反映组织中的总镁含量。研究表明,即使血清镁浓度正常,细胞内镁含量也可能耗竭。因此,仅考虑血液中的镁含量,而不考虑膳食镁摄入量和尿液丢失量,可能会低估临床上的镁缺乏症。
低镁血症患者常出现低钾血症。顽固性低钾血症通常与镁缺乏有关,只有在镁水平恢复正常后,低钾血症才能得到有效纠正。镁缺乏会促进钾从集合管分泌,进一步加剧钾流失。细胞内镁水平降低会抑制Na+–K+–ATP酶活性,并增加肾外髓质钾(ROMK)通道开放,导致更多钾从肾脏流失。镁和钾之间的相互作用还涉及激活钠-氯共转运体(NCC),从而促进钠重吸收。镁缺乏通过一种名为神经前体细胞发育下调4-2(NEDD4-2)的E3泛素蛋白连接酶降低NCC丰度,并通过低钾血症阻止NCC激活。NCC持续下调可增强低镁血症状态下远端Na+转运,导致尿钾排出增多和低钾血症。
多种类型药物,如抗生素、利尿剂、生物制剂、免疫抑制剂、质子泵抑制剂(PPI)和化疗药物,都可能导致镁丢失和低镁血症(图3)。长期使用PPI会导致约20%患者出现镁缺乏,这些影响与剂量相关。PPI会减少肠道对镁的吸收,并导致肠腔pH值和肠道微生物群发生变化。只有部分患者会出现PPI引起的低镁血症,这可能与PPI治疗剂量和持续时间、膳食镁摄入量、与其他导致镁丢失药物同时使用以及肠道微生物群状况有关。据报道,口服菊粉可以通过增加胃肠道吸收,改善PPI引起的低镁血症患者的血清镁水平。
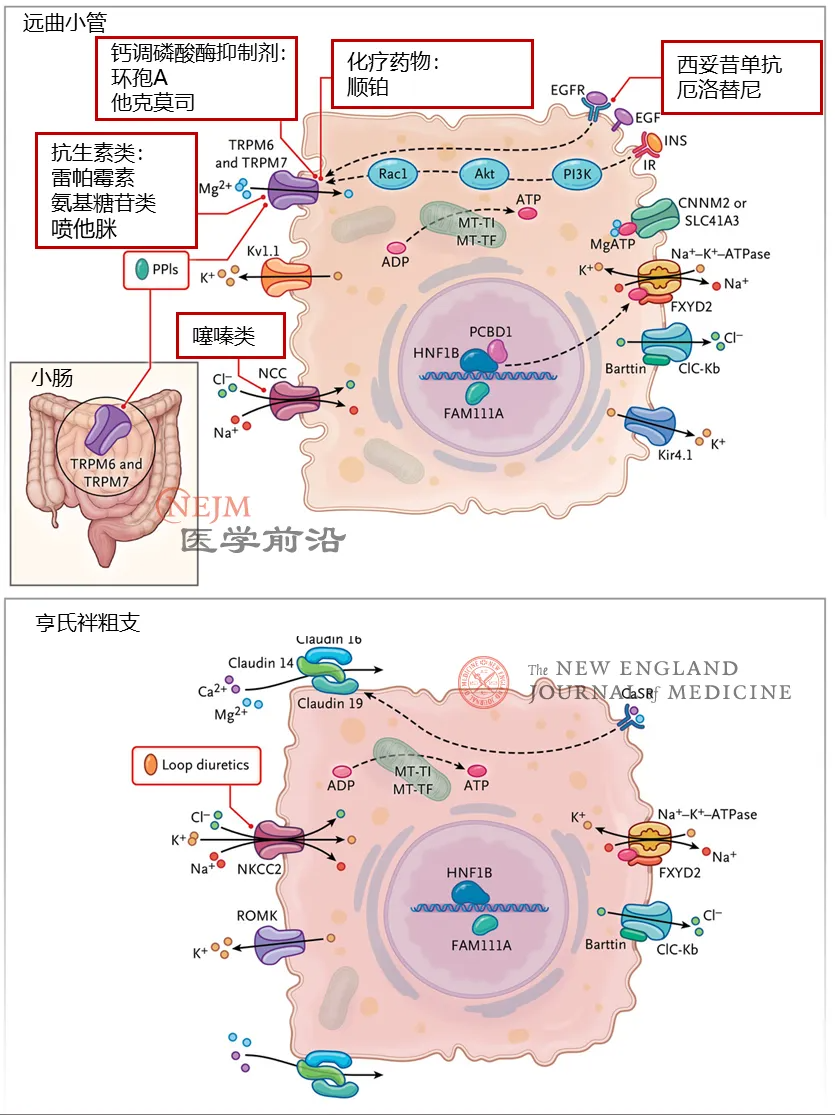
图3. 镁稳态以及低镁血症的体征和症状以及治疗方法。
低镁血症常见于2型糖尿病患者。肾脏丢失镁过多和血液中白蛋白结合增加可能是低镁血症的原因。胰岛素可激活远曲小管中的TRPM6活性,因此,胰岛素抵抗会导致肾脏对镁重吸收减少和尿镁增多。
镁平衡异常还与心血管疾病有关。低镁血症易导致心电不稳定和心律不齐,包括心房颤动、尖端扭转型室性心动过速和长QT综合征。在血管系统中,低镁血症与内皮功能障碍、血管收缩、血管张力增加和血管纤维化有关—这些都是高血压的特征。前述血管效应涉及TRPM7活性改变和血管细胞中镁离子流入受损,这在醛固酮增多症中尤为明显。
先兆子痫和子痫是妊娠期高血压的并发症,其特征是滋养层异常侵入、内皮功能障碍和血管炎症。这两种情况下的血镁水平可能正常或降低。虽然先兆子痫和子痫的病因尚不清楚,但静脉注射镁治疗是有益的,可以预防或减轻并发症。镁的保护作用归因于钙通道阻滞和血管扩张。然而,其他因素也与此有关,包括FMS样酪氨酸激酶1和内皮素水平降低、氧化应激减少、脑NMDA受体抑制、促炎介质生成减少以及TRPM6和TRPM7下调。
对编码镁转运通路及其调节因子基因的致病性变异进行鉴定,已推定约80%的家族性低镁血症的遗传病因。大多数低镁血症的遗传因素会影响远曲小管对镁的重吸收。TRPM6和TRPM7亚基的突变会导致低镁血症和继发性低钙血症。此类患者的低钙血症可归因于甲状旁腺功能减退,而甲状旁腺功能减退则是由于甲状旁腺细胞内镁含量低,从而影响PTH分泌。EGF和EGFR致病性变异会导致TRPM6活性降低,从而引发低镁血症和肾脏丢失镁。CNNM2的致病性变异会导致低镁血症、癫痫发作和认知缺陷。CNNM2在远曲小管中表达,并调节细胞基底侧镁排出,但分子机制尚不清楚。
Gitelman综合征主要是由NCC突变引起的钠排泄障碍。然而,Gitelman综合征患者会出现低镁血症、低钾血症和代谢性碱中毒。该综合征中低镁血症的原因尚不清楚,临床前数据表明,当NCC出现缺陷时,远曲小管萎缩可能导致镁重吸收减少。
异亮氨酸和苯丙氨酸的线粒体转运RNA(MT-TI和MT-TF)的致病性变异与Gitelman综合征样低镁血症有关。这些突变与线粒体电子传递链复合物4的活性降低有关。因此,细胞ATP生成受损可能会降低细胞基底侧Na+–K+–ATP酶的活性并抑制NCC。
NCC活性受其细胞内N端结构域的磷酸化调节。一种被称为“钾开关”的机制可以解释细胞外低钾如何导致基底膜超极化和NCC激活。编码基底侧钾通道(KCNJ10和KCNJ16)、氯通道(CLCNKB和BSND)和Na+–K+–ATP酶复合体(ATP1A1和FXYD2)的钾开关基因突变会导致与Gitelman综合征相似的表型,包括低镁血症。HNF1B突变患者易患肾畸形、肾囊肿和青少年发病型糖尿病(MODY5)。
编码紧密连接蛋白16和19的CLDN16和CLDN19突变会导致家族性低镁血症、高钙尿症和肾钙盐沉着症。亨氏袢升支中的claudin 16和19形成阳离子选择性孔,允许钙和镁从细胞旁重吸收。携带CLDN19致病变异的患者与携带CLDN16变异的患者可通过是否存在眼部缺陷来区分。在亨氏袢升支中中,细胞外二价阳离子转运的主要调节因子是PTH和钙离子感应受体。RAS相关GTP结合D(RRAGD)的致病性变异已被确认为肾小管病变表型的病因,该表型与家族性低镁血症、高钙尿症和肾钙盐沉着症(FHHNC)表型合并扩张型心肌病类似。
尽管镁与多种疾病相关,但只有少数几种情况需要使用镁作为首选治疗剂。这些情况包括尖端扭转型室性心动过速、急性哮喘加重以及先兆子痫或子痫。对于β受体阻滞剂耐药的尖端扭转型室性心动过速,镁被视为治疗的一种选择。对于重症哮喘急性发作且对初始强化治疗反应不佳的患者,以及患有危及生命的哮喘的患者,临床指南建议静脉注射硫酸镁。雾化硫酸镁与吸入β2-受体激动剂和异丙托溴铵联合使用,可能对肺功能有额外益处,并可能减少或缩短住院时间。镁对哮喘的益处可能与其通过钙通道阻滞作用于支气管平滑肌,从而扩张支气管有关。
镁是临床诊疗中一种重要但往往被忽视的电解质。它很少作为常规电解质进行检测。低镁血症通常没有症状。尽管调节体内镁平衡的确切机制尚不明确,但对肾脏处理镁的机制研究已经取得了进展。许多药物都会导致低镁血症。低镁血症在住院患者中很常见,也是导致ICU住院时间延长的危险因素。低镁血症应通过有机盐制剂形式的来纠正。尽管关于镁在健康和疾病中的作用还有很多待解之谜,但该领域已经取得诸多进展,临床医生应该更加重视镁在临床医学中的重要性。